糖尿病是一组以高血糖为特征的代谢性疾病,典型病例可出现多尿、多饮、多食、消瘦等表现,即“三多一少”症状,糖尿病一旦控制不好会引发并发症,如神经病变、肾病、视网膜病变和心血管疾病。两种最常见的糖尿病类型是1型糖尿病(T1D)和2型糖尿病(T2D)。1型糖尿病是一种自身免疫性疾病,可导致胰岛中产生胰岛素的胰腺β细胞的破坏。在糖尿病患者中,2型糖尿病约占90%以上。2型糖尿病主要由胰岛素抵抗和胰岛素分泌不足引起。动物模型在疾病病理生理学和靶点鉴定以及评价新的治疗药物和体内治疗中发挥着关键作用。本文概述了1型和2型糖尿病小鼠及大鼠模型,包括NOD小鼠、BB大鼠、LEW-IDDM大鼠、AKITA小鼠、ZDF大鼠、化学诱导糖尿病模型等。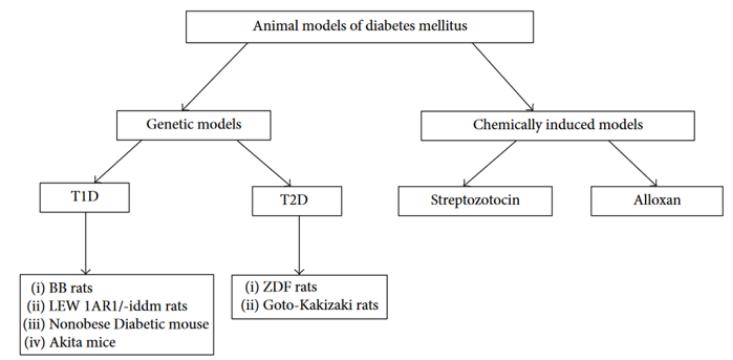
Figure 1. 糖尿病动物模型总结
1型糖尿病的主要特征是β细胞的自身免疫性破坏,导致胰岛素生产的缺乏。在动物模型中,这种胰岛素产生的缺乏是通过多种不同的机制来实现的,从化学破坏β细胞到自发发展为自身免疫性糖尿病的啮齿动物。
自发性模型
1型糖尿病最常用的自发性模型是NOD小鼠、BB大鼠和LEW-IDDM大鼠。其中,NOD小鼠作为自发性模型仍占主导地位。NOD小鼠是1974年在日本大阪的Shionogi实验室开发的。该模型的小鼠因免疫细胞激活而导致胰岛β细胞被袭击破坏,进而呈现出糖尿病的临床表现。NOD小鼠在3-4周龄左右出现胰岛素炎,明显的糖尿病的发病通常直到10-14周时才明显出现,糖尿病可以发展到30周。糖尿病在雌鼠中发病率为60%至90%,而在雄鼠的发病率为10%至30%。BB大鼠来源于远系繁殖的Wistar大鼠。雄性和雌性的发病率相似。大约90%的大鼠在8至16周龄时会患上糖尿病。糖尿病表型相当严重,大鼠需要胰岛素治疗才能生存。LEW-IDDM大鼠:该大鼠在一个MHC单倍型的同源Lewis大鼠群体中自发产生。这些大鼠表现出胰岛素炎,并在大约8-9周时表现出明显的糖尿病。糖尿病的发病率约为60%左右,雌雄鼠发病率相同。与NOD小鼠和BB大鼠相比,LEW-IDDM大鼠没有表现出其他自身免疫性疾病,在明显的糖尿病发作后也存活良好,因此可以用于研究糖尿病并发症。
遗传诱导的胰岛素依赖性糖尿病模型
AKITA小鼠起源于C57BL/6NSlc小鼠,该小鼠的INS2基因发生了自发突变,阻止了胰岛素原的正确加工,导致错误折叠的蛋白质超载和内质网应激[1]。AKITA杂合子小鼠从3-4周龄开始发生严重的1型糖尿病,其特征是高血糖、低胰岛素血症、多尿和多饮。未经处理的纯合子很少能存活超过12周。该模型通常用于研究胰岛内质网应激的潜在缓解剂,也是2型糖尿病的病理模型之一。Akita小鼠无需处理即可维持糖尿病状态长达11个月时间。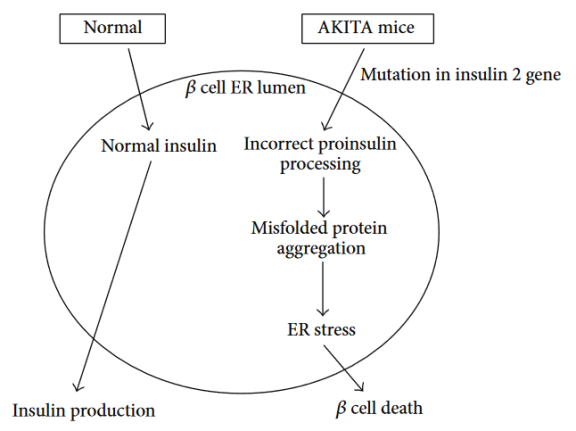
Figure 2. Akita小鼠发病原理
化学诱导模型
化学诱导糖尿病模型通常在实验开始前5-7天左右诱导,以确保稳定的高血糖症。两种主要的化合物被用于诱发糖尿病:链脲佐菌素(STZ)或四氧嘧啶(ALX)[2]。化学诱导糖尿病适用于测试药物或治疗方法。STZ是由消色性链霉菌合成的。通过腹腔注射或尾静脉注射给药后,它通过Glut-2转运体进入胰腺β细胞,导致DNA烷基化[3]。随后PARP的激活导致NAD+的消耗,细胞ATP的减少,随后胰岛素的产生被抑制。此外,STZ是一种自由基的来源,也可能导致DNA损伤和细胞死亡。STZ倾向于作为单次高剂量或多次低剂量给予。造模方法:注射前用0.05mol/L柠檬酸(pH4.5)配成2%的STZ溶液,新鲜使用。大鼠糖尿病STZ的单次刺激剂量为40-65mg/kg(ip或iv)。小鼠糖尿病STZ的单次刺激常用剂量为100-200mg/kg(ip或iv)。ALX糖尿病作用主要归因于β细胞的快速摄取和自由基的形成,而β细胞对自由基的防御机制较差。四氧嘧啶被还原为二尿酸,然后再氧化回四氧嘧啶,创造一个超氧化物自由基产生的氧化还原循环,超氧自由基经过分解形成过氧化氢,然后高活性的羟基自由基导致β细胞DNA断裂。造模方法:一般配成1%-3%浓度即可使用。给药剂量依动物及给药途径不同而异(均需临用前配置),大鼠150-200 mg/kg(ip),40-60 mg/kg(iv);小鼠200 mg/kg(ip),85-100 mg/kg(iv)。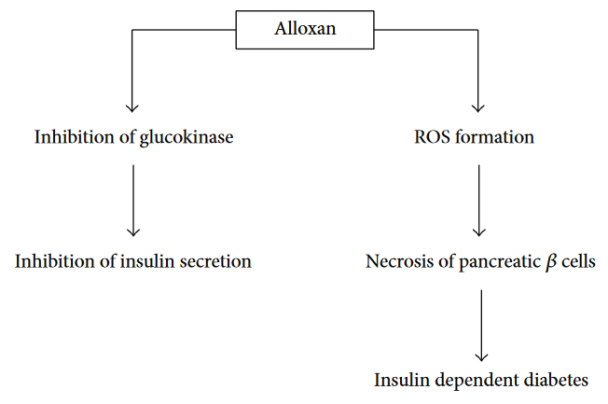
Figure 3. 四氧嘧啶的主要病理作用
单基因模型
虽然人类肥胖很少由单基因突变引起,但单基因肥胖模型在2型糖尿病研究中普遍使用。最广泛使用的肥胖单基因模型是在瘦素信号传递方面存在缺陷的。瘦素会诱发饱腹感,因此,在这些动物中缺乏功能性瘦素会导致暴饮暴食和肥胖。这些模型包括Lepob/ob小鼠、Leprdb/db小鼠和Zucker糖尿病脂肪大鼠(ZDF)。Lepob/ob小鼠:Lepob/ob小鼠是一种严重肥胖的模型,小鼠4号染色体的瘦素受体基因发生缺陷,从而导致自发性2型糖尿病。体重在2周龄时开始增加,小鼠出现高胰岛素血症。到4周时,明显出现高血糖,血糖浓度持续上升,在3-5个月达到峰值,之后随着小鼠年龄的增长而下降。Leprdb/db小鼠:该小鼠是瘦素受体基因的常染色体隐性突变。表现为暴饮暴食、肥胖、高胰岛素血症和高血糖症。肥胖从3-4周龄开始明显,高胰岛素血症在大约2周龄时开始明显,在4-8周龄时出现高血糖。最常用的背景是C57BLKS/J,它们在几个月后发生酮症,寿命相对较短。Zucker脂肪大鼠和Zucker糖尿病脂肪大鼠:1961年Merck M-strain和Sherman杂交后发现了Zucker大鼠。它们瘦素受体突变,可以诱导暴饮暴食,并且在4周龄左右变得肥胖。这些大鼠患有高胰岛素血症、高脂血症和高血压。这些大鼠的糖耐量受损,但不表现明显的糖尿病,但该品系的突变产生了一个具有明显糖尿病表型的Zucker糖尿病脂肪大鼠,这些大鼠比Zucker脂肪大鼠更瘦,但有更严重的胰岛素抵抗。其特征是在8周龄左右时最初出现高胰岛素血症,随后胰岛素水平下降。雄鼠通常在8-10周左右发生糖尿病,但雌鼠不会发展明显的糖尿病。
多基因模型
KK-AY小鼠:KK小鼠是具有和人的肥胖性糖尿病相似特征的动物模型,是K.Kondo在1941年应用Kasukabe小鼠原种群培育得来的具有先天性遗传缺陷性小鼠。因KK小鼠没有显著的表型特征,研究者将突变毛色基因(ay)转入小鼠体内,形成KK-AY小鼠[4]。ay基因可以影响小鼠的毛发,更重要的是其可以引发代谢紊乱,导致小鼠出现明显的肥胖和高血糖等表现,而雄性小鼠的表现更为明显,此种动物模型广泛用于T2DM的实验模型。OLETF大鼠:OLETF大鼠来自1984年在Long Evans大鼠远系群体中发现的一只自发性糖尿病大鼠。选择性育种导致OLETF大鼠患有轻度肥胖和晚发性高血糖(18周后)。糖尿病是由雄性遗传而来的。胰岛经历了三个阶段的组织学改变:在早期阶段(6-20周龄),可见细胞浸润和变性,20到40周为增生阶段,最后阶段的特征是胰岛纤维化,并被结缔组织所取代。OLETF大鼠也表现出肾脏并发症。NZO小鼠:NZO小鼠是一种通过选择性繁殖产生多基因肥胖模型。NZO小鼠在9-12周龄时就出现了高瘦素血症,瘦素抵抗导致暴饮暴食和肥胖。血糖浓度升高,并表现出糖耐量受损。随着年龄的增长而恶化,大约50%的雄鼠会患上糖尿病。胰岛在3-6个月大时出现增生和肥大,但再大一些月龄出现胰岛β细胞丢失。
诱导肥胖模型
此模型建立在高脂肪日粮(HFD)饲养的基础上,通常是使用含有50%-60%脂肪的食物持续饲喂小鼠,饲喂长短取决于研究所需模型病变的严重程度[5]。HFD饲养可导致肥胖、高胰岛素血症和由于胰岛补偿不足而导致的葡萄糖稳态改变。一般情况下,在60%的HFD喂食1周后,小鼠表现出葡萄糖耐量受损和胰岛素分泌增加,2-4周后体重明显增加,8-12周时口服/静脉/腹腔葡萄糖耐量实验和葡萄糖刺激胰岛素释放实验的结果将出现明显变化,体重将在16-20周达到最大,最终体重通常比野生型重20%~30%。这类小鼠模型通常表现为肥胖、高血压、高血糖和高胰岛素血症。HFD小鼠能很好地模拟人类T2DM的进展,并表现出胰岛素抵抗、胰岛β细胞增多及β细胞功能破坏等特征。
上面描述了各种1型和2型糖尿病的动物模型,每一种都有各自的特点。这些糖尿病模型可以用于多种不同的研究目的,包括药理学测试、遗传学研究和了解疾病机制。模型的选择将取决于研究的目的。在1型糖尿病中,选择动物模型的主要决定因素是是否需要一个自身免疫模型。在不同的模型中,发病的时间和可预测性是不同的。在2型糖尿病中,需要特别考虑的是高血糖的机制以及这是否与你的研究有关。这些机制可能包括胰岛素抵抗和/或胰岛β细胞衰竭。此外,应注意的是,一些糖尿病动物模型不会出现糖尿病并发症,因此如果研究糖尿病并发症应当谨慎选择。
参考文献:
[1] King AJ. The use of animal models in diabetes research. British journal of pharmacology 2012, 166(3): 877-894.
[2] Al-Awar A, Kupai K, Veszelka M, Szűcs G, Attieh Z, Murlasits Z, et al. Experimental Diabetes Mellitus in Different Animal Models. Journal of diabetes research 2016, 2016: 9051426.
[3] Furman BL. Streptozotocin-Induced Diabetic Models in Mice and Rats. Current protocols 2021, 1(4): e78.
[4] Rees DA, Alcolado JC. Animal models of diabetes mellitus. Diabetic medicine : a journal of the British Diabetic Association 2005, 22(4): 359-370.
[5] King A, Bowe J. Animal models for diabetes: Understanding the pathogenesis and finding new treatments. Biochemical pharmacology 2016, 99: 1-10.
本文转自小张聊科研

